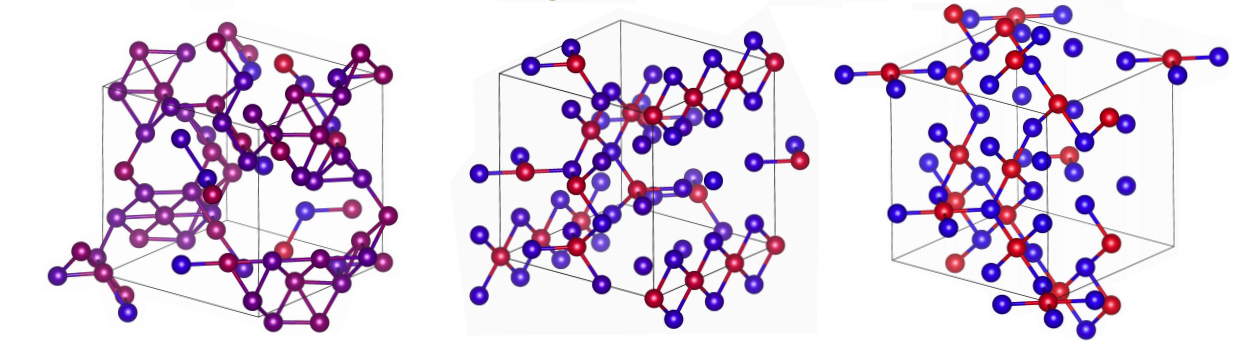
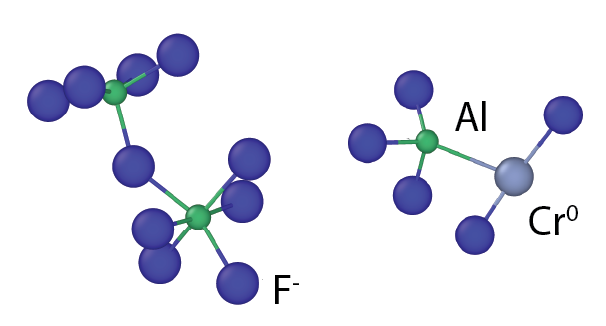
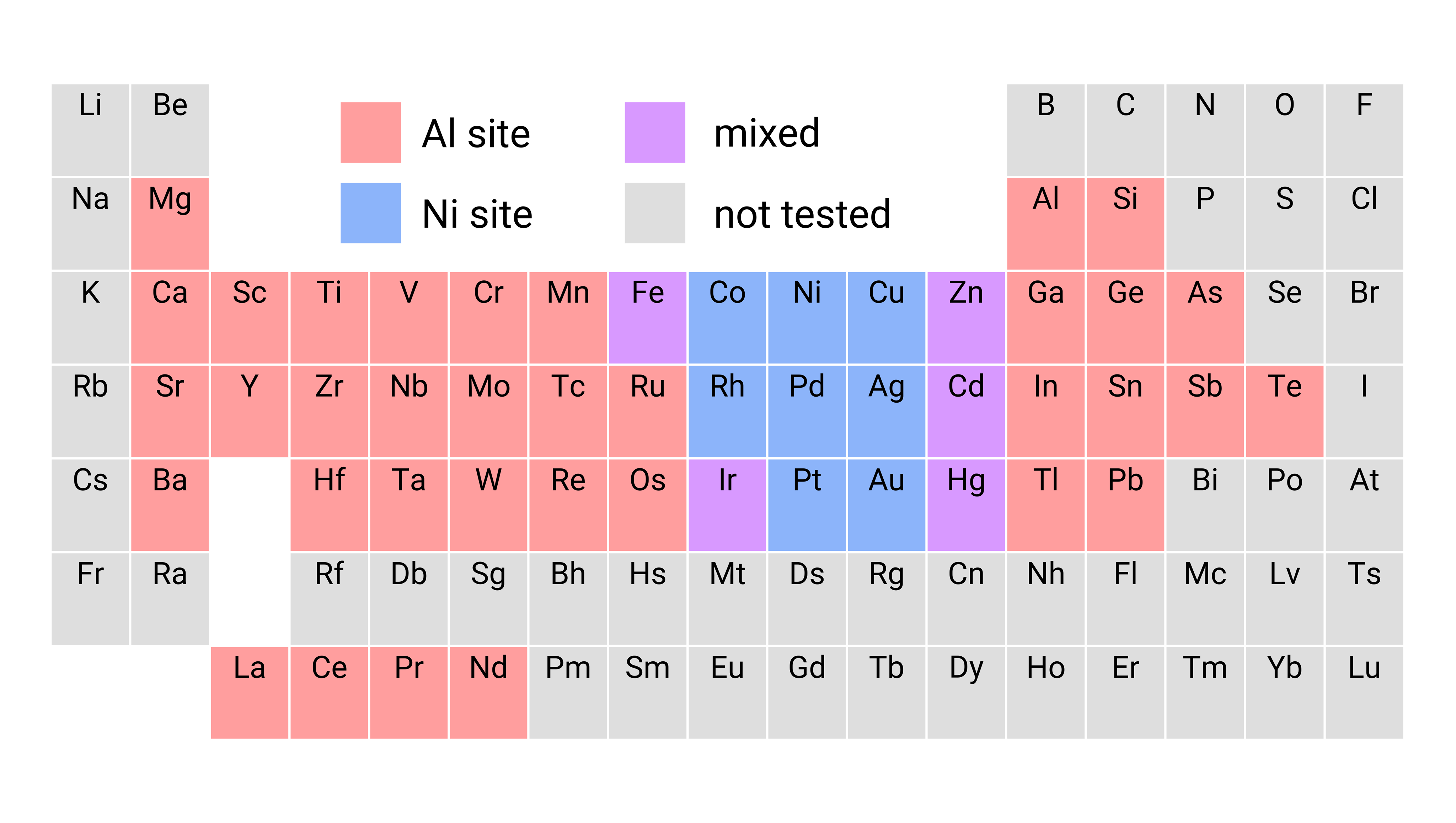
Welcome to the Asta Group
We leverage state-of-the-art computational techniques and resources to study advanced materials for the next generation of engineering technologies. Our current research area focuses include advanced structural alloys, functional semiconductors, Generation IV nuclear reactor materials, and code development. We are located in the Department of Materials Science and Engineering at the University of California, Berkeley.

Recent News
-
December 2023
Enze graduates with his Ph.D. in Materials Science and Engineering and will start as a Lecturer at Stanford University in the new year!
-
September 2023
Wenqing passes the qualifying exam to advance to Ph.D. candidacy!
-
August 2023
Yuan passes the preliminary exam – congratulations, Yuan!
-
July 2023
The group says farewell to David Olmsted who retires after many years as a Staff Scientist with the Asta group.
-
May 2023
Madelyn and Nathan pass their qualifying exams and advance to Ph.D. candidacy!
Anas graduates with his Ph.D. in Materials Science and Engineering and begins work as an NRC Postdoctoral Fellow at NIST.
Flynn graduates with his Ph.D. in Applied Science and Technology and begins work at Lawrence Livermore National Lab as a Postdoctoral Fellow.
Bella graduates with a B.S. in Materials Science in Engineering and will begin graduate school at Stanford.
-
March 2023
The group welcomes Zhao Fan as a new Postdoctoral Researcher, co-advised by Michael Whittaker and Piotr Pawel Zarzycki.
Nick graduates with his Ph.D. and begins work at Apple as an Applied Scientist.
Enze receives the Materials Science and Engineering Graduate Student and Inclusion Award for 2022-2023!